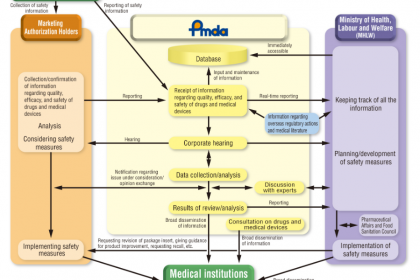
在全球医疗器械监管体系中,日本凭借严谨的分类标准、清晰的监管层级、完善的法律法规及专业化的审评机构,构建了覆盖医疗器械全生命周期的监管与注册框架。日本医疗器械的管理核心由卫生劳动和福利部(Ministry of Health, Labor and Welfare,MHLW,即日本厚生省)统筹,其管辖的独立行政法人药品和医疗器械综合机构(Pharmaceuticals and Medical Devices Agency,PMDA)承担核心技术审评、复核等职能。本文系统梳理日本医疗器械的分类标准、核心监管机构(PMDA与MHLW)职责、相关法律法规、全流程注册要求,深入解析各环节核心要点,为医疗器械企业拓展日本市场、合规开展注册申报及相关从业者提供、、系统的行业参考(本文所指医疗器械不含体外诊断试剂)。
一、核心监管机构解析:PMDA与MHLW的职能分工与关联
日本药品和医疗器械的监管体系以“MHLW统筹管控、PMDA专业执行”为核心,两者权责清晰、协同联动,共同保障医疗器械的、有效与合规性,明确界定了日本医疗器械监管的核心架构。
(一)厚生劳动省(MHLW):统筹监管的核心主管部门
MHLW全称为Ministry of Health, Labor and Welfare(卫生劳动和福利部),即日本传统意义上的厚生省,是日本医疗器械监管的主管部门,主要承担统筹规划、法规制定、最终审批等宏观管控职能,具体职责包括:
- 统筹负责日本药品、医疗器械的管理,制定监管战略与发展规划;
- 审批发布医疗器械相关法律法规、标准规范,明确监管要求与实施细则;
- 对PMDA的工作进行监督与指导,审定PMDA提交的医疗器械审评结果、认证结论,最终作出承认、批准等决策;
- 负责医疗器械上市后监管的宏观统筹,协调处理重大风险与违规事件。
(二)PMDA:专业执行的技术审评机构
PMDA全称为Pharmaceuticals and Medical Devices Agency(独立行政法人药品和医疗器械综合机构),是MHLW管辖下的独立行政法人,专注于医疗器械、药品等的技术审评、管控等专业工作,是日本医疗器械监管体系的核心执行机构,具体职责包括:
- 医疗器械上市前技术审评,包括新器械、改良器械、仿制器械的审评复核,为MHLW的最终承认提供专业技术支撑;
- 开展医疗器械相关研究工作,完善审评标准与技术规范,参与监管体系优化;
- 承担医疗器械上市后对策,跟踪产品使用,处置隐患与不良事件;
- 负责健康损害救济相关工作,规范救济流程,保障医疗器械使用者的合法权益;
- 开展制造商注册审核、质量管理体系审核等,规范企业生产经营行为;
- 为企业提供临床试验、注册申报等相关咨询服务,指导企业合规申报。
(三)PMDA与MHLW的核心关联
PMDA隶属于MHLW,接受MHLW的监督与指导,两者形成“宏观统筹+专业执行”的协同机制:PMDA负责所有医疗器械的技术复核、审评、审核等专业工作,提出审评意见与建议;MHLW基于PMDA的专业意见,负责最终的审批、承认、法规发布等宏观管控,确保监管工作的专业性与性统一。
二、日本医疗器械相关法律法规体系
日本药品、医疗器械管理法律法规体系层次清晰,分为法律、政令/法令、告示/省令三个层级,层层衔接、明确具体,为医疗器械分类、监管、注册等工作提供了明确的法律依据,确保监管工作有法可依、有章可循。
1. 法律:由日本议会批准通过,是医疗器械监管的层级法规,具有法律效力,明确医疗器械监管的核心原则、整体框架与核心要求,指导后续政令、告示的制定。
2. 政令/法令:由日本政府内阁批准通过,是对法律的细化与补充,明确法律条款的具体实施细则,界定各相关部门、机构的权责分工,确保法律能够有效落地执行。
3. 告示/省令:由MHLW(厚生省)大臣批准通过,是医疗器械监管的具体实施标准,涵盖医疗器械分类基准、注册流程、审评标准、质量管理要求等具体内容,是企业合规申报、监管机构开展工作的直接依据(如MHLW Ordinances NO.69关于质量管理体系的要求)。
其中,《药品和医疗器械法案》(Pharmaceutical and Medical Device Act, PMD Act)是日本医疗器械监管的核心法律,明确了医疗器械制造商注册、产品注册、质量管理等核心要求,是所有医疗器械相关活动的根本遵循。
三、日本医疗器械分类标准:基于风险等级的分级体系(结合JMDN编码)
日本医疗器械的分类核心遵循“风险等级适配监管强度”原则,结合日本医疗器械术语集(Japanese Medical Device Nomenclature, JMDN)编码,明确界定器械分类与注册登记路径,依据产品风险等级由低到高,分为Ⅰ类(一般)、Ⅱ类(管制)、Ⅲ类(高度管制)、Ⅳ类(高度管制)四个等级,各类别界定清晰、适用范围明确,为后续差异化监管与注册申报提供基础依据。
1. 一般医疗器械(Ⅰ类):风险等级,定义为“即使发生不良事件,对人体的风险也极低”的产品,如手术刀等。此类产品结构简单、性高,无需复杂的性验证即可保障使用,监管与注册方式以地方政府备案为主,无实质性审查。
2. 管理医疗器械(Ⅱ类,管制医疗设备):风险等级中等,定义为“即使发生不良事件,对人体的风险也比较低”的产品,如电子内窥镜、消化器官用导管等。此类产品结构与功能相对复杂,存在一定潜在风险,需通过第三方认证机构(RCB)负责审查,完成认证后即可合规上市。
3. 高度管理医疗器械(Ⅲ类,高度管制医疗设备):风险等级较高,定义为“在发生不良事件时,对人体的风险比较高”的产品,如透析器、人工骨骼、人工呼吸器、心脏血管用球囊导管等。此类产品技术难度较高,直接影响人体健康,需由PMDA进行专业技术审评,经MHLW(厚生省)承认后,方可上市。
4. 高度管理医疗器械(Ⅳ类,高度管制医疗设备):风险等级,定义为“对患者的侵入性高、在发生不良事件的情况下有可能直接导致生命危险”的产品,如起搏器、人工心脏、支架等。此类产品性要求,直接关系人体生命,需由PMDA进行、严格的技术审评,经MHLW(厚生省)承认后,方可上市。
截至2018年4月1日,日本MHLW(厚生省)已制定946个认证基准、45个承认基准和9个审查指导原则,结合JMDN编码,为各类医疗器械的分类界定、监管实施、注册申报提供了明确的标准依据,确保分类与监管的科学性、统一性。
四、日本医疗器械注册全流程详解(合规申报核心路径)
日本医疗器械注册流程规范、环节清晰,核心遵循“分类确定—主体任命—制造商注册—体系审核—申报审批”的逻辑,不同风险等级产品的注册要求有所差异,整体流程可分为四大核心步骤,确保注册工作合规、落地。
(一)步:确定产品分类(核心前提)
产品分类是日本医疗器械注册的核心前提,需依据JMDN编码及风险等级,明确产品所属类别(Ⅰ类、Ⅱ类、Ⅲ类、Ⅳ类),不同类别对应不同的注册路径、审查机构与监管要求。企业需先完成产品分类界定,确保后续注册工作贴合产品特性,避免因分类偏差导致注册延误或违规。
(二)第二步:任命MAH/D-MAH(上市许可持有人)
根据日本医疗器械监管要求,所有类别医疗器械在开展上市前申请或审批前,必须先任命Marketing Authorized Holder(MAH,日本上市许可持有人)或Designated Marketing Authorization(D-MAH,指定上市许可持有人),负责管理产品上市申报、后续合规管控等相关事宜,具体要求如下:
- MAH:企业需拿到对应类别产品的MAH执照后,方可提出具体产品的上市申请,负责产品上市后的管控、投诉处理等;
- D-MAH:针对外国企业(在日本无办事处),必须任命一名在日本持有营业执照的D-MAH,主要负责协调货物放行给外国企业的经销商,处理产品投诉、警戒信息等相关事宜,是外国企业进入日本医疗器械市场的核心衔接主体。
(三)第三步:进行制造商注册(MR/FMR)
根据PMD Act(《药品和医疗器械法案》)要求,医疗器械产品投放到日本市场前,制造商必须完成注册登记,获得相应注册证书,该证书是提交医疗器械产品注册申请的必备条件,具体分为两种类型:
1. 制造商注册(MR):适用于日本本国制造商,需向当地地方当局提交制造商注册(MR)申请,审核通过后获得MR证书,明确生产场所合规性;
2. 外国制造商注册(FMR):适用于外国制造商,需向PMDA提交外国制造商注册(FMR)申请,审核通过后获得FMR证书,确保外国制造商的生产场所、质量管理符合日本监管要求。
MR与FMR证书均为产品注册申报的前置条件,企业需在提出产品注册申请前,完成制造商注册并取得对应证书。
(四)第四步:质量管理体系审核(J-GMP/QMS)
质量管理体系合规是日本医疗器械注册的核心要求之一,核心遵循J-GMP(日本药品生产质量管理规范,MHLW Ordinances NO.69)及QMS(质量管理体系)相关标准,不同类别产品的审核要求差异化明确:
- Ⅰ类医疗器械:无需进行J-GMP审核,仅需完成地方政府备案即可;
- Ⅱ类医疗器械:由注册认证机构(RCB)负责开展J-GMP审核,审核通过后,方可进入后续注册申报环节;
- Ⅱ类(除特殊控制外)、Ⅲ类、Ⅳ类医疗器械:由PMDA负责开展QMS审核,严格核查企业质量管理体系的合规性、有效性,审核通过后,方可进入技术审评环节。
(五)补充说明:不同类别产品的后续申报与审批
完成上述四大核心步骤后,企业需根据产品类别,提交相应的注册申报资料,进入最终的审评与审批环节:
- Ⅰ类医疗器械:完成地方政府备案,无实质性审查,备案完成后即可上市;
- Ⅱ类医疗器械:向RCB提交申报资料,由RCB完成审查,审查通过后即可上市;
- Ⅲ类、Ⅳ类医疗器械:向PMDA提交申报资料,由PMDA完成技术审评,审评通过后报MHLW(厚生省)承认,获得承认后即可上市。
五、核心总结
日本医疗器械监管与注册体系以“风险分级、管控、合规”为核心,形成了“MHLW统筹、PMDA执行、多机构协同”的完善架构。MHLW作为主管部门,负责法规制定与最终审批;PMDA作为专业执行机构,承担技术审评、体系审核等核心工作;结合JMDN编码的分类标准、层次清晰的法律法规,以及规范的注册流程,构建了覆盖医疗器械全生命周期的监管与注册生态。
对于医疗器械企业而言,把握产品分类、明确PMDA与MHLW的职能分工、严格遵循注册全流程要求,是合规进入日本市场、实现长效发展的关键。本文系统梳理的分类标准、监管机构、法律法规及注册流程,可为相关企业与从业者提供的参考,助力其完成合规申报与运营。